12 KiB
Lithium - ein HPC Cluster für die Fachhochschule Graubünden
Hardware
Für Berechnungen in der Forschung und Lehre kann die Fachhochschule Graubünden seit Herbst 2023 auf einen High Performance Computing (HPC) Cluster zurückgreifen. Dieser wurde aufgebaut um die Lehre und Forschung bei grossen Datenmengen und rechenintensiven Aufgaben zu Untersützen und die Studierenden mit den Prinzipien des verteilten Rechnens bekannt zu machen.
Der HPC Cluster entspricht in seinem Aufbau einem Beowulf Cluster. Dieser nutzt spezialisierte Softwarebibliotheken aus dem OpenHPC Repository für das verteilte Rechnen. Zur Verwaltung des Clusters verwenden wir die Software Warewulf in einer sogenannten 'stateless' Konfiguration. Dadurch ist sichergestellt, dass die Knoten (einzelne Cluster Computer) softwaremässig identisch installiert und konfiguriert sind, was verhindert, dass es zu Versionsdifferenzen auf den einzelen Knoten kommt.
Der Cluster basiert auf einer von der Firma Sanitas gespendeten und vom DAViS/CDS auf Open Source Komponenten umgenutzten Oracle Big Data Applicance X5-2L mit sechs Nodes (Knoten).
Ein Node besteht aus den folgenden Komponenten
| 2 x CPU | Xeon E5-2699 v3 @ 2.30GHz |
|---|---|
| RAM | 128 GB DDR-4 |
| Disks | 12 x 14 TB 7200 RPM SAS |
und sieht so aus:

Der gesamte Cluster mit Netzwerkequipment so:

Für die Intranode-Kommunikation beim Rechnen über RDMA (zum Beispiel MPI) kommt ein High Performance Interconnect auf Basis von Infiniband QDR (40 Gbps) zum Einsatz. Für die Verwaltung der Nodes (SSH, TFTP etc.) steht ein dediziertes 10 GB Ethernet Netz zur Verfügung.
Im Hardwarevergleich steht unser Rechner hier:
| Notebook | Lithium | CSCS Alps | |
|---|---|---|---|
| Physische Cores | 4 Cores | 216 Cores | 65536 Cores |
| Memory (RAM) | 16 GB | 768 GB | ? |
| Speicherplatz | 1 TB | 504 TB | 10 PB |
| Energieverbrauch | 60 W | 5-6 KW | 5-10 MW |
| Interconnect | 1 Gb/s | 40 Gb/s | 200 Gb/s |
Das bedeutet, dass wir hardwaremässig sehr viel kleiner sind als Cluster im Bereich der Top500. Auf der Anwenderseite sind wir jedoch sehr ähnlich ausgerüstet wie die grossen HPC Forschungscluster. Auf Lithium findet man daher unter anderem folgende Komponenten:
- Slurm für das Job Scheduling
- BeeGFS (paralleles Filesystem) auf dem Scratchlaufwerk /scratch
- MPICH für das verteilte Rechnen
- Warewulf für die Clusteradministration
Im gesamten sind über 300 HPC spezifische Softwarepakete installiert.
Architektur
Der Fachhochschulcluster besteht aus sechs Knoten (engl. Nodes). Dabei sind die einzelnen Nodes über ein Low Latency, High Bandwith RDMA Netzwerk miteinander verbunden. Dieses Netzwerk kommt hauptsächlich während einer Berechnung zum Einsatz.
Einer der sechs Nodes ist als "Master", respektive "Headnode" oder "Loginnode" konfiguriert. Dieser ist während einer Berechnung für Koordinations- und Steueraufgaben zuständig und wird zudem zur Verwaltung des Clusters verwendet. Auf diesem Node wird prinzipiell nicht gerechnet.
Die eigentliche Rechenaufgabe wird für die parallele Abarbeitung auf die Computenodes verteilt. Nach Abschluss der Berechnung werden die Resultate entweder automatisch durch das verwendete Skript oder manuell über das /scratch oder Homelaufwerk zurück an den Masternode geschickt.
Der Fachhochschule Cluster ist wie folgt aufgebaut:
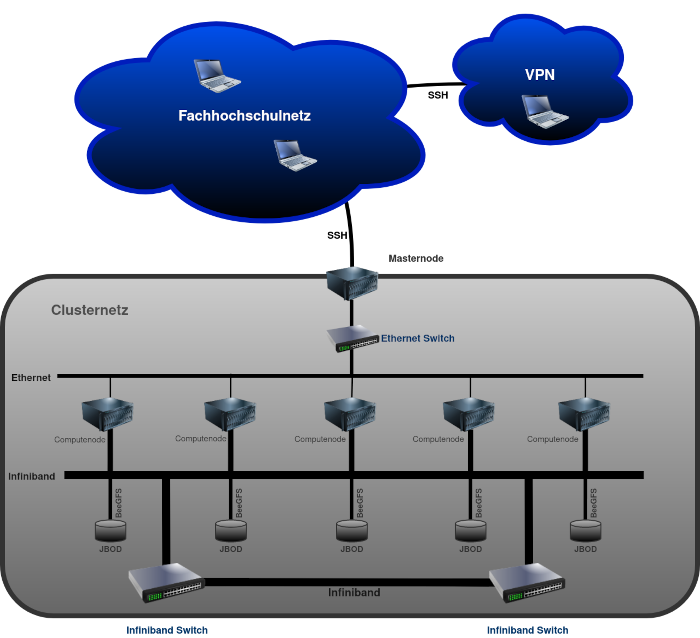
Zugriff auf den Cluster
Auf den Cluster kann nur mit SSH zugegriffen werden. Momentan können sich alle Mitarbeitenden des SIIs und alle Studierenden des Studiengangs CDS darauf einloggen. Für den Login braucht es einen SSH Client und einen FHGR Account. Der Zugriff mit SSH funktioniert so:
ssh <FH Benutzername>@lithium.fhgr.ch
Falls Du den Cluster verwenden möchtest, jedoch nicht zum SII oder CDS Studiengang gehörst, melde dich bitte beim DAViS Admin (thomas.keller@fhgr.ch).
Starten der Computenodes
Um Energie zu sparen, werden unbenutzte Computenodes automatisch heruntergefahren. Um eine Berechnung zu starten, ist es daher notwendig, dass die Computenodes gebootet sind und sich im Slurmstate 'IDLE' befinden. Sollte der Status mit dem Befehl sinfo --all als 'down' angezeigt werden, dann müssen die Computenodes gestartet werden. Das Starten aller Nodes ist mit dem folgenden Befehl möglich:
sudo /usr/local/bin/start-computeNodes.bash
Nach einigen Minuten, sollte der Status des Clusters von 'DOWN' auf 'IDLE' wechseln. Sollte einer der folgenden Status anzeigt werden 'IDLE*' (mit Stern), 'DRAINED', 'DRAINING', 'FAIL', 'FAILING', 'FUTURE', 'POWER_DOWN', 'UNKNOWN', 'RESERVED' dann informiere bitte, den DAViS Administrator.
Job Scheduler und Partitionen
Da ein Cluster auch ein Mehrbenutzersystem ist, können dessen Resourcen nicht jederzeit frei verwendete werden. Daher kommt ein Jobscheduler zum Einsatz, der Rechenjobs mit den zur Verfügung stehenden Ressourcen möchglichst optimal zur Ausführung bringt. Auf lithium.fhgr.ch werden die Jobs durch Slurm verwaltet.
Grundsätzlich folgt Slurm auf Lithium dem FIFO mit Backfill Prinzip. Vereinfacht gesagt bedeutet das, dass der erste eingereichte Job zuerst abgearbeitet wird. Weiter wird die Ressourcennutzung durch sogenannte Partitionen eingeschränkt. Diese bestimmen wie lange ein Benutzer einen Slurm-Job ausführen darf. Momentan sind die folgenden Partitionen konfiguriert:
| Anwendergruppe / Partitionen | Debug | Studierende | Mitarbeitende |
|---|---|---|---|
| Studierende | 5 Min | 3 h | kein Zugriff |
| Mitarbeitende | 5 Min | kein Zugriff | 12 h |
Eine Berechnung die länger als die durch die Partition vorgegebene Zeit läuft wird abgebrochen. Diese Limite ist dazu da, damit ein Benutzer nicht irrtümlich oder absichtlich den Cluster für eine unbegrenzte Zeit blockieren kann. Daher empfiehlt es sich dringend, im Skript sogenannte 'Checkpoints' zu implementieren. Wie Checkpoints im Falle von Tensorflow oder Keras implementiert werden, findest Du hier. Checkpoints schützen übrigens auch vor einem Zitverlust bei einem Stromausfall.
Falls Du deutlich mehr als die oben erwähnten Zeitspannen für eine Berechnung brauchst, melde dich beim DAViS Admin.
Clusterfilesystem und Homeverzeichnis
Auf dem Cluster wird das Homeverzeichnis von jedem Benutzer automatisch mit NFS auf die Computenodes exportiert. Das heisst, dass auf jedem Node, egal ob Master oder Computenode die Daten des Homeverzeichnisses zur Verfügung stehen. Übrigens: auf deinem Homeverzeichnis steht jedem Anwender 80 GB Speicherplatz zur Verfügung. Wieviel Speicherplatz Du bereits belegt hast, kannst Du mit dem Befehl quota -fs / ausfindig machen.
Des weiteren gibt es unter dem Verzeichnis /scratch ein geshartes Filesystem mit einer Grösse von ca. 500 TB. Hier kommt BeeGFS, ein paralleles Filesysteme zum Einsatz das Speicherplatz von allen Computenodes unter dem Mountpoint (Verzeichnis) /scratch vereint. Daher kann es nur genutzt werden wenn alle Computenodes gestartet sind. Wie das Homverzeichnis ist auch das /scratch Laufwerk von allen Nodes zugreifbar.
Bitte beachte, dass Dateien unter dem Verzeichnis /scratch von allen Clusterbenutzern gelesen aber nur vom Dateieigentümer verändert werden können.
Stromverbrauch und Energiesparmodus
Mit dem Befehl
sudo /usr/local/bin/get-powerUsage.bash
kann der momentane Stromverbrauch des Clusters in Erfahrung gebracht werden. Dabei ist der Stromverbrauch des High Performance Transports (in unserem Falle Infiniband) nicht mit eingerechnet.
Der Energiesparmodus wird aktiviert, falls der letzte Slurm Job vor mehr als 90 Minuten abgeschlossen wurde oder die Clusternodes erst kürzlich gebootet wurden. Solange Slurmjobs ausgeführt werden, werden die belegeten Clusternodes nicht in einen Energiestparmodus versetzt oder ausgeschaltet.
Installation von zusätzlicher Software
Viele Machinlearning und Deeplearning Frameworks oder Python Module können über Anaconda, Miniconda oder Apptainer installiert werden. Für Anhaltspunkte wie das gemacht werden kann, siehe die Anleitung Softwareinstalltionen auf den Workstations
Erste Schritte auf dem Cluster
Mit dem Befehl srun kann ein Slurmjob auf dem Cluster ausgeführt werden. Als simples Beispiel dient hier der Befehl 'hostname', der den Namen des lokalen Rechners anzeigt.
srun -p students -n $((36*5)) -N5 hostname
Mit der Option -p wird die Partition ausgewählt, im obigen Fall die 'students' Partition. Mit der Option -n teilen wir Slurm mit, wieviele parallele Tasks (Prozesse) wir ausführen wollen. Da wir pro Computenode 36 physische Cores haben (Hyperthreading ist deaktiviert) können wir auf dem Cluster maximal 180 Prozesse starten. Die Option -N teilt Slurm mit wieviele Computenodes für die Berechnung verwendet werden sollen. Im obigen Fall sollten wir daher genau 180 Zeilen Output erhalten.
Mit dem Befehl squeue kann angezeigt werden welche Jobs momentan ausgeführt werden und welche Jobs auf Ressourcen warten.
Für nicht-interaktive und länger laufende Jobs ist es sinnvoll sbatch zu verwenden. Damit muss nicht gewartet werden bis der Cluster frei wird, sondern der Jobscheduler informiert den Anwender per Mail sobald das Skript zur Ausführung kommt.
Dafür muss ein Shellskript geschrieben werden das einerseits einen Abschnitt enthält mit Informationen für Slurm (diese Befehle sind mit #SBATCH gekenntzeichnet) andererseits einen Abschnitt der den Befehl zum Starten der Berechnungen enthält. Dies könnte zum Beispiel so aussehen:
#!/bin/bash
#SBATCH --nodes=5 ## Number of computenodes
#SBATCH --output="slurm-%j.out" ## Logfile
#SBATCH --time=240:00 ## Time limit. Should be equal or smaller than the partitions time limit
#SBATCH --job-name="test-mpi" ## Job name. This will be used in naming directories for the job.
#SBATCH --partition=staff ## Partition to launch job in
#SBATCH --cpus-per-task=1 ## The number of threads the code will use
#SBATCH --ntasks-per-node=1 ## Number of tasks (processes/threads) to run
#SBATCH --mail-type=BEGIN,END ## Sends a Email when Job starts and when the Job ends
#SBATCH --mail-user=<mail address> ## Mail Address
# Load an environment
module load py3-mpi4py
# Execute the python script and pass the argument '90'
srun python3 my-mpiProg.py 90
MPI Hello World
Der Cluster unterstützt das Message Passing Interface welches das parallele und verteilte Rechnen in einem Programm erlaubt. Um MPI zu nutzen, muss der Programmcode angepasst werden. Ein Hello World mit C und MPI sieht zum Beispiel so aus:
#include <stdio.h>
#include "mpi.h"
int main(int argc, char *argv[]) {
char hostname[1024];
hostname[1023] = '\0';
gethostname(hostname, 1023);
int ierr;
ierr = MPI_Init(&argc, &argv);
printf( "Hello from %s\n", hostname);
ierr = MPI_Finalize( );
exit(ierr);
}
Da das Programm die MPICH Impelmentierung nutzen soll, laden wir zuerste das entsprechende Module
module load intel mpich
Danach kompilieren wir das Program mit dem Befehl:
mpicc ./hello_world_mpich.c -o hello_world_mpich
Falls noch nicht erfolgt, sollten sptätestns vor dem Ausführen, die Cluster Resourcen mit dem Befehl salloc alloziert werden. Danach kann das Program mit dem Befehl
mpirun /home/kellerthomas@edu.local/testcluster/hello_world_mpich
aufgerufen werden. Dabei