13 KiB
GPU Workstations
Hardware
Für Berechnungen im Studium stehen an der Fachhochschule Graubünden verschiedene Workstations zur Verfügung. Eine Übersicht über die Maschinen findest Du
Zugriff
Der Zugriff erfolgt über SSH aus den Netzen der FH Graubünden (WLAN, VPN). Wie eine SSH Verbindung eingerichtet wird, ist hier beschrieben.
Architektur
Alle Workstations für das Studium sind nach dem folgenden Muster aufgebaut:
| Was | Beschreibung |
|---|---|
| Login auf den Workstations | Für das Login mit SSH verwendest Du deinen FH Graubünden Benutzernamen und Passwort |
| Homeverzeichnis und Quota | Dein Homeverzeichnis ist auf 80 GB Speicherplatz beschränkt. Welche Möglichkeiten Du hast falls dein Homeverzeichnis voll belegt ist, siehst Du hier |
Speicherplatz unter /scratch |
Für temporäre Dateien die die Quota von 80 GB überschreiten, kann unter /scratch Speicherplatz verwendet werden. In diesem Verzeichnis bitte keine vertrauliche Daten ablegen |
| Slurm für den Zugriff auf die GPUs | Die GPUs können nur mit Slurm verwendet werden. Wie das funktioniert steht im Abschnitt Job Scheduler und Partitionen |
| Installation zusätzlicher Software | Python Module, Entwicklungs Bibliotheken etc. können über die Software Anaconda/Miniconda oder Pip installiert werden. Wie das am Beispiel von Tensorflow funktioniert ist im Dokument |
| Container | Für die Arbeit mit Containern ist die Software Apptainer installiert. Wie mit Apptainer Software installiert werden kann (auch für Software die Rootrechte benötigt) ist im Dokument |
Job Scheduler und Partitionen
Da alle Workstation an der FH Mehrbenutzersysteme sind, können deren Resourcen (wie die GPUs) nicht jederzeit frei verwendete werden. Daher kommt ein Jobscheduler zum Einsatz, der Rechenjobs mit den zur Verfügung stehenden Ressourcen möchglichst optimal zur Ausführung bringt. Daher musst Du bei einer Berechnung mit GPUs zwingend verwenden.
Grundsätzlich folgt Slurm auf den Workstations dem FIFO mit Backfill Prinzip. Vereinfacht gesagt bedeutet das, dass der erste eingereichte Job zuerst abgearbeitet wird. Weiter wird die Ressourcennutzung durch sogenannte Partitionen eingeschränkt. Diese bestimmen wie lange ein Benutzer einen Slurm-Job ausführen darf. Die möglichen Partitionen kannst Du dir mit dem Befehl sinfo anzeigen lassen. Typischerweise gibt es für die Studierenden die foglenden zwei Partition:
| Partition | Zweck | Maximale Dauer |
|---|---|---|
| Debug | Zum Testen und Experimentieren | 5 Min |
| Students | Für lange laufende Berechnungen | 7 Tage |
| Staff | Für lange laufende Berechnungen | 14 Tage |
Slurm weiss nicht, wann dein Job sich beendet hat. Da die wenigsten Berechnungen im Studium sieben Tage oder mehr dauern, solltest Du deinen Slurmjob nachdem deine Berechnung abgeschlossen, ist von Hand beenden (Mit squeue (Zeigt die Job ID an) und scancel beenden). Besser noch, Du gibts beim Starten des Batchjobs die ungefähre Laufzeit deiner Berechnung an. Dies kannst Du mit dem Parameter --time= angeben. Zum Beispiel --time=1:30:00 für einen Slurmjob der maximal 1 Stunde und 30 Minuten laufen soll. Damit Slurm deine Berechnung nicht vorzeitig abbricht addiere zur Schätzung der Laufzeit deiner Berechnung noch eine zeitliche Sicherheitsmarge dazu.
Falls deine Berechnung weniger als sieben Tage benögtigt, sind dir deine Mitstudierenden sicherlich dankbar, wenn Du den Slurmjob beendest sobald deine Berechnung abgeschlossen ist. Ansonsten bleibt die GPU für 7 Tage blockiert.
Eine Berechnung die länger als die durch die Partition vorgegebene Zeit läuft wird abgebrochen. Diese Limite ist dazu da, damit ein Benutzer nicht irrtümlich oder absichtlich den Cluster für eine unbegrenzte Zeit blockieren kann. Daher empfiehlt es sich dringend, im Skript sogenannte 'Checkpoints' zu implementieren. Wie Checkpoints im Falle von Tensorflow oder Keras implementiert werden, findest Du hier. Checkpoints schützen übrigens auch vor einem Datenverlust der Berechnung bei einem Stromausfall oder Diskausfall.
Falls Du deutlich mehr als die oben erwähnten Zeitspannen für eine Berechnung brauchst oder ein Slurmjob die GPUs für sehr lange Zeit blockiert, melde dich bitte beim .
Slurm Commands
Slurm auf den Workstations wird zwingend benötigt, falls deine Berechnung auf der GPU ausgeführt werden soll. Jobs, die nur auf der CPU rechnen, müssen Slurm nicht verwenden. Bei sehr intensiver und lange anhaltender CPU Belegung, empfehlen wir jedoch eine Nutzung von Slurm, damit eine parallel laufende GPU Berechnung nicht gestört wird indem die CPU oder Memory Resourcen komplett belegt werden.
Welche Hardware wir auf dem Rechner zur Verfügung haben, können wir mit dem Befehl
sinfo -o "%N %c %m %G" | column -t
in Erfahrung bringen.
Mit dem Befehl srun kann ein Slurmjob auf der Workstation ausgeführt werden. Als simples Beispiel berechnen wir hier die Primfaktoren einer grossen Zahl
srun -G a100:1 -p debug -n 64 factor 1234567890123456789012345678901234567890
Mit der Option -p wird die Partition ausgewählt, im obigen Fall die 'debug' Partition. Mit der Option -n teilen wir Slurm mit, wieviele parallele Tasks (Prozesse) wir ausführen wollen. Da wir pro Computenode 64 hyperthreading Cores zur Verfügung haben, können wir den Parameter -n auf maximal 64 Tasks setzen. Die Option -G a100:1 fordert eine Nvidia A100 GPU für die Berechnung an. Sobald die Computerresource frei ist, wird der Befehl srun ausgeführt und es werden auf der CPU 64 Prozesse gestartet. Sobald die Berechnung abgeschlossen ist, wird das Ergebnis auf der Kommandozeile ausgegeben.
Übrigens: im obigen Befehl, ist die Option -G a100:1 nicht nötig und nur als Beispiel aufgeführt, da der Befehl factor die GPU nicht nutzen kann.
Für nicht-interaktive und länger laufende Jobs ist es sinnvoll den Befehl sbatch zu verwenden. Damit muss nicht gewartet werden bis die Workstation frei wird, sondern der Jobscheduler übernimmt das Skript und bringt es zur Ausführung sobald die Hardwareresourcen frei sind. An welcher Reihe sich mein Job in der Jobqueue befindet, kann mit dem Befehl
squeue angezeigt werden.
Ein weiterer Vorteil von sbatch ist, dass der Job beim Unterbruch der Netzwerkverbindung nicht abgebrochen wird wie zum Beispiel mit srun. Wenn Du srun oder andere interaktive Befehle ausführen möchtest, die sehr lange laufen, dann empfiehlt es sich zu nutzen, damit bei einem Netzwerkunterbruch dein Befehl nicht abgebrochen wird.
Für sbatch muss ein Shellskript geschrieben werden das einerseits einen Abschnitt enthält mit Informationen für Slurm (diese Befehle sind mit #SBATCH gekenntzeichnet) andererseits einen Abschnitt der den Befehl zum Starten der Berechnungen enthält. Dies könnte zum Beispiel so aussehen:
#!/bin/bash
#SBATCH --output="slurm-%j.out" ## Im Verzeichnis aus dem sbatch aufgerufen wird, wird ein Logfile mit dem Namen slurm-[Jobid].out erstellt.
#SBATCH --error="slurm-%j.err" ## Ähnlich wie --output. Jedoch ein Log für Fehlermeldungen.
#SBATCH --time=1:30:00 ## Zeitlimite. Diese sollte gleich oder kleiner der Partitions Zeitlimite sein. In diesem Fall ist diese auf 1 Stunde und 30 Minuten gesetzt.
#SBATCH --job-name="Mein Test" ## Job Name.
#SBATCH --partition=students ## Partitionsname. Die zur Verfügung stehenden Partitionen können mit dem Befehl sinfo angezeigt werden
#SBATCH --cpus-per-task=1 ## Die Anzahl Threads die Slurm starten soll
#SBATCH --ntasks-per-node=64 ## Die Anzahl Prozesse die gestartet werden sollen
# Ausführen des effektiven Befehls in der Shell. Bei einer Berechnung würde hier typischerweise ein Python Skript aufgerufen
srun -G a100:1 -p students -n 64 factor 1234567890123456789012345678901234567890
Ob das Skript nun aktiv ist (oder einfach nur hängt ohne etwas zu machen) ist nicht immer ganz eindeutig festzustellen. Jedoch sieht man mit dem Befehl top oder sudo nvtop ob das gestartete Skript CPU oder GPU Resourcen verwendet. Weiter kann in den Logfiles die im sbatch Skript angegeben wurden, nachgeschaut werden ob es Fehler bei der Skriptausführung gegeben hat. Zum Beispiel für den Job 101 mit dem Befehl cat slurm-101*.
Es ist auch möglich eine interaktive Slurm Session zu nutzen. Dazu kann zum Beispiel der folgende Befehl am Workstation-Prompt eingegeben werden:
salloc -p students --time=00:01:00
Danach können wir unsere Befehle interaktiv in der Konsole aufrufen.
Um die Auslastung der GPUs anzuzeigen kann der Befehl
sudo nvtop
aufgerufen werden.
Ein Beispiel einer Berechnung
Im folgenden Beispiel verwenden wir das MNIST Datenset um ein einfaches Tensorflow Modell zu trainieren. Wr verwenden in diesem Beispiel einen Apptainer Container. Du kannst Tensorflow aber auch mit Anaconda/Miniconda installieren. Siehe dazu Installation Tensorflow
Login auf einer der Workstations
ssh <username>@mercury.fhgr.ch
Einrichten einer Execution Environment für Tensorflow mit Apptainer
mkdir -p ~/build-apptainer/sandboxes/ && cd ~/build-apptainer
apptainer pull tensorflow-2.16.1-gpu.sif docker://tensorflow/tensorflow:2.16.1-gpu
apptainer build --sandbox sandboxes/tensorflow ./tensorflow-2.16.1-gpu.sif
apptainer exec --fakeroot --writable sandboxes/tensorflow/ apt-get update
apptainer exec --writable --fakeroot sandboxes/tensorflow/ apt install -y nvidia-profiler
Starten eines Slurmjobs
Um auf die GPUs der Workstation zuzugreifen, brauchen wir zwingend eine Slurm Session. Da unser Programm nur für eine GPU ausgelegt ist, fordern wir auch nur eine GPU mit Slurm an (die zweite GPU kann von einem weiteren Benutzer für eine gleichzeitig laufende Berechnung angefordert werden):
salloc -p students --time=2:00:00 -G a100:1 --ntasks=32 --mem-per-cpu=7G
Sobald unser Slurm Job an der Reihe ist um ausgeführt zu werden (siehe squeue), können wir mit den folgenden Befehlen testen ob wir Zugriff auf die GPU haben:
apptainer shell --nv "${HOME}/build-apptainer/tensorflow-2.16.1-gpu.sif"
python3 <<- EOF
import tensorflow as tf
print(tf.config.list_physical_devices('GPU')[0])
EOF
exit
In der letzten Ausgabezeile unseres Skripts sollte im Falle eines Erfolgs nun
PhysicalDevice(name='/physical_device:GPU:0', device_type='GPU')
stehen. Falls anstatt der obigen Meldung ein Index Error erscheint (IndexError: list index out of range), hat der Zugriff auf die GPU nicht geklappt.
Als nächstes berechnen wir ein kleines . Dazu kopieren wir den folgenden Code in eine Datei im Homeverzeichnis mit dem Namen ml.py:
# https://www.tensorflow.org/tutorials/quickstart/beginner
import tensorflow as tf
print("TensorFlow version:", tf.__version__)
mnist = tf.keras.datasets.mnist
(x_train, y_train), (x_test, y_test) = mnist.load_data()
x_train, x_test = x_train / 255.0, x_test / 255.0
model = tf.keras.models.Sequential([
tf.keras.layers.Flatten(input_shape=(28, 28)),
tf.keras.layers.Dense(128, activation='relu'),
tf.keras.layers.Dropout(0.2),
tf.keras.layers.Dense(10)
])
predictions = model(x_train[:1]).numpy()
predictions
tf.nn.softmax(predictions).numpy()
loss_fn = tf.keras.losses.SparseCategoricalCrossentropy(from_logits=True)
loss_fn(y_train[:1], predictions).numpy()
model.compile(optimizer='adam',
loss=loss_fn,
metrics=['accuracy'])
model.fit(x_train, y_train, epochs=5)
model.evaluate(x_test, y_test, verbose=2)
probability_model = tf.keras.Sequential([
model,
tf.keras.layers.Softmax()
])
probability_model(x_test[:5])
Danach starten wir unseren Tensorflow Container mit dem Befehl
`apptainer shell --nv "${HOME}/build-apptainer/tensorflow-2.16.1-gpu.sif"
Der Kommandozeilen Prompt wechselt nun sein Aussehen zu Apptainer>
Da unser Skript (ml.py) im Homeverzeichnis liegt, können wir unser ml.py Skript mit dem Befehl
python3 "$HOME/ml.py"
aufstarten. Eine erfolgreiche Berechnung sieht so aus:
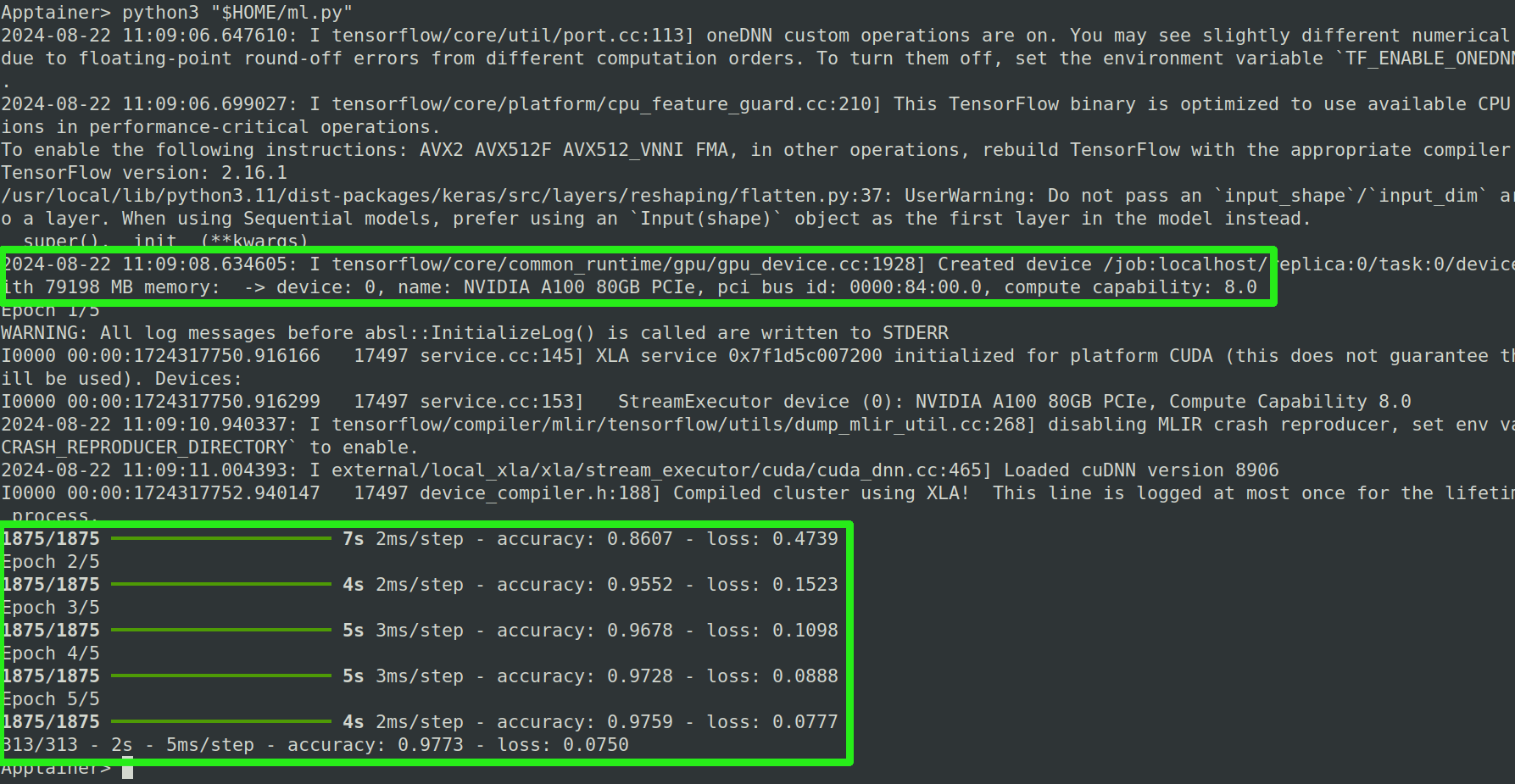
Falls während der Initialisierung des Skripts der Fehler failed call to cuInit: CUDA_ERROR_NO_DEVICE: no CUDA-capable device is detected auftaucht, wurde die Berechnung auf der CPU ausgeführt und nicht auf der GPU. In diesem Fall überprüfe nochmals ob der Container tatsächlich in einer Slurm Session gestartet wurde (und nicht in einer zweiten Konsole ohne Slurm) und ob die Slurm Session immer noch aktiv ist (Mit dem Befehl squeue).